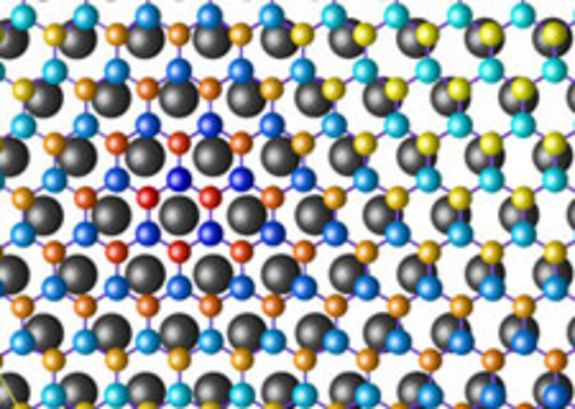
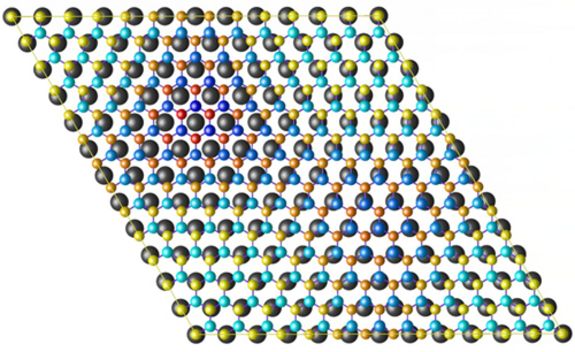

Die neuen Erkenntnisse wurden erst unlängst in einer März-Ausgabe (PRL 98, 106802, (2007)) des wissenschaftlichen Fachjournals "Physical Review Letters" publiziert sowie im Virtual Journal of Nanoscience and Technology zitiert. Die Wissenschafter vom Institut für Materialchemie beschäftigten sich im Rahmen eines Projektes mit der Selbstorganisation auf Oberflächenstrukturen, im konkreten Fall mit Bornitrid auf metallischen Rhodiumschichten. Bornitrid entspricht einem Graphitgitter, in dem man Kohlenstoff abwechselnd durch Bor und Stickstoff ersetzt hat. Bisher wurde vermutet, dass sich beim Abscheiden auf Rhodium Bornitrid in Form einer Doppelschicht bildet, die geordnete Löcher mit einem Durchmesser von etwa 2 Nanometer aufweisen. Schwarz und seinen Mitarbeitern gelang es die Struktur mit Hilfe von Computersimulationen nachzurechnen.
Die ursprüngliche Annahme von zwei Schichten konnte daraufhin widerlegt werden. Schwarz: "Es handelt sich um nur eine Schicht von Bornitrid auf Rh-Metall. Die regelmäßige Anordnung in diesem hexagonalen Gitter mit einer Periodizität von 3,2 nm kommt dadurch zustande, dass die Länge von 12 Bornitrideinheiten mit 13 Rh-Gitterabständen übereinstimmen. Dadurch binden Bor und Stickstoff in manchen Oberflächenbereichen besser oder schlechter an die darunter liegende Metallschicht, sodass sich die Atome einmal näher am Metall und einmal weiter weg anordnen. Dies führt letztlich zu einer selbst geordneten Nanostruktur, die für Mikroelektronikanwendungen wichtig werden kann."
Die Untersuchungen in Zusammenarbeit mit der Universität Zürich liefern wichtige Erkenntnisse für die Konzeption von Mikrobauteilen, die beispielsweise für Speichermedien verwendet werden. Ge-rade die Nanotechnologie bietet hier einen reichen wissenschaftlichen Fundus. Die Simulationen wurden auf einem Cluster aus PCs mit insgesamt 72 Doppelprozessoren gerechnet. Ein seit den 80er Jahren an der TU Wien entwickeltes Computerprogramm mit dem Namen "WIEN2k" schuf die nötigen Voraussetzungen für die Berechnung. "Das Computerprogramm haben wir ständig weiterentwickelt. Früher, als ich meine Dissertation geschrieben habe, konnten wir zwei Atome pro Elementarzelle (pro kleinster Einheit) berechnen. Jetzt liegen wir bei über 1.000 Atomen."